What a great run going on at JPR right now! Every time I go to finish my post on TeaCup, I end up distracted by something new that has just dropped that I could really use right now!

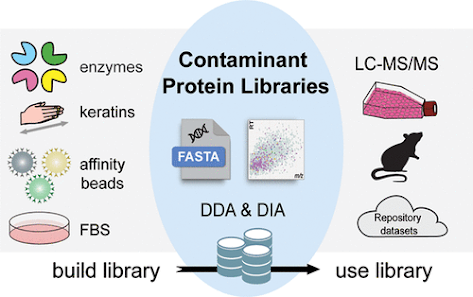
This group rightfully points out that while we’ve got some great FASTA databases out there for contaminants, we don’t really use FASTAs for DIA. They also put in a load of work comparing the MaxQuant contaminant database to cRAP to their in-house developed contaminants.
They go the extra mile in using a lot of software in their pipeline and in the tools that they’ve made publicly available in just about every way you can think of.
There is a nice instructional Github here.
All the original RAW files for the contaminant library generation is here.
The data was generated using an HF-X which is probably worth keeping in mind if you’re using other hardware. For example, I’ve found that while tools like Prosit do an amazing job of matching fragment intensities for Orbitrap spectra, they don’t match quite as well for TIMMY devices, particularly for ions of higher m/z where fragmentation and isolation efficiency of the latter isn’t all that great due to the lack of a robust CE normalization algorithm and quadrupole limitations, respectively.
There are some really cool examples in this paper of matches that look really good until you compare the data to the new contaminant spectral libraries they’ve generated and then — whoops — that’s definitely a peptide from LysC!
Turns out that if the only thing you think of at G.W. is the scammy old super computer thing that skims hundreds of millions of dollars off the federal government every year despite not one published study in about a decade, think again, there is a forward thinking proteomics/multiomics group there worth keeping an eye on!
Thanks for sharing. I read many of your blog posts, cool, your blog is very good. https://accounts.binance.com/ru/register?ref=V3MG69RO